近日����,中國科學技術大學李微雪教授結合物理啟發的可解釋機器學習算法與第一性原理計算,解決了一個多相催化研究中長期存在的關于催化結構敏感性難題���。研究成果于近日以“Structure Sensitivity of Metal Catalysts Revealed by Interpretable Machine Learning and First-principles Calculations”為題發表于《美國化學會》期刊(J. Am. Chem. Soc.)。
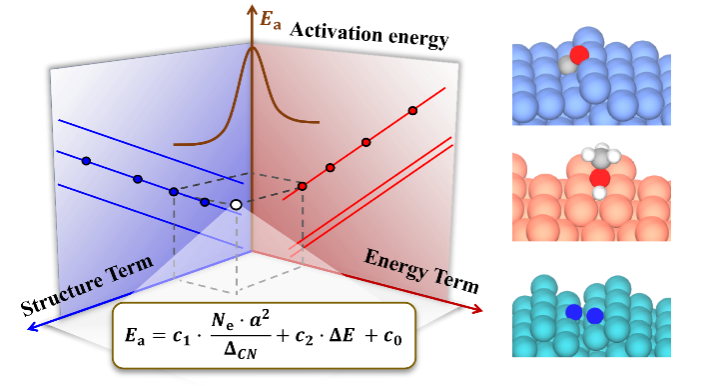
催化反應活性位及其結構敏感性是多相催化研究中最為重要的基本概念之一���。盡管近年來研究取得了很大進展,但由于影響因素眾多并橫跨多個空間和時間尺度��,如何在原子尺度上確定催化反應的活性位及其結構敏感性,依然是催化材料理性設計中所面臨的一大挑戰�����。舉例來說�,活化能與反應熱之間的Br?nsted–Evans–Polanyi(BEP)關系以及不同分子吸附能之間的線性標度律,長期以來被視為催化反應機理和催化優化設計的最重要的基本研究框架��。但是��,由于BEP關系和標度律中缺乏催化劑幾何結構和化學組分的明確信息�����,這使它們原則上無法描述催化結構敏感性�����,從而極大限制了催化劑的優化設計研究���。
機器學習方法在多相催化研究中發揮著日益重要的作用���,并被應用到催化劑的結構敏感性研究中���。但迄今為止大多數研究都屬于端到端的“黑盒子”研究�,研究結果缺乏很好的物理可解釋性��。物理上具有清晰的可解釋性�����、明確包含催化劑的幾何結構和化學組分、并能準確預測催化反應能壘的解析關系����,目前仍然亟待建立�����。另外,由于催化反應能壘的計算主要通過高精度��、高成本的密度泛函理論來完成��,系統的理論數據也較為匱乏。因此,經常需要參考不同的數據源�����,數據源的多樣性所帶來的挑戰也需要采取合適的機器學習算法��。
面對這一問題�����,在本研究工作中,作者基于物理啟發的可解釋多任務學習符號回歸和包含多樣性的第一性原理計算數據集,在領域知識和化學直覺的基礎上��,建立了一個簡潔���、物理圖像清晰的描述符�。該描述符由催化劑的結構項和催化反應的能量項兩部分組成,可用于準確預測各種分子在不同組分和結構金屬催化劑上的活化能壘。其中,新建立的結構項由催化劑的拓撲配位不飽和度、價電子和晶格常數三個變量組成�����,借此成功破解了金屬催化劑的結構敏感性問題���,并突顯了數據驅動理論模型的透明度(“白盒子”研究)在構建催化物理模型方面的重要性���。
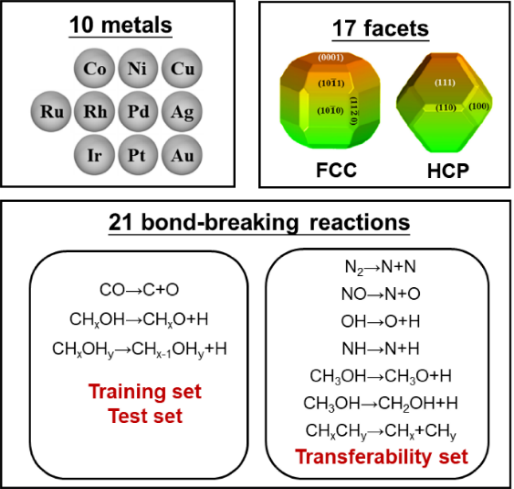
圖1 涵蓋了多種金屬�����、晶面����、晶相和反應的第一性原理計算數據集
在具體機器學習建模過程中,該工作提出了幾點關鍵策略和思路。首先��,為保障數據集的多樣性�,構建了一套涵蓋了21種不同化學鍵斷鍵能壘����、10種過渡金屬催化劑、2種不同晶相和17種不同晶面的較大多源數據集(圖1)��。其次�����,根據領域知識和化學直覺���,推斷表面能與活化能應該存在很強關聯�����,而表面能又與表面懸掛鍵或配位不飽和度存在關聯。在綜合考慮表面暴露各種不同配位原子貢獻的情況下,作者定義了一個新的�����、純粹反映催化劑結構特征的物理量:拓撲配位不飽和度ΔCN
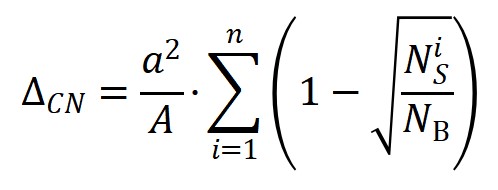
圖2a顯示��,催化反應的活化能與拓撲配位不飽和度的相關性明顯優于表面能和傳統BEP中的反應熱�����。將這一拓撲配位不飽和度,與反應熱以及催化劑的其它基本原子參數作為物理特征一起放到相應的機器學習研究中�。第三�,為保障數據驅動模型的可解釋性���、準確性和通用性等��,考慮到多源數據可能含有的非自洽性,以及不同分子之間的差異難以顯式描述等情景��,該工作采用了多任務學習符號回歸策略進行機器學習建模���。
機器學習結果最終給出的分子活化能最佳模型如下:
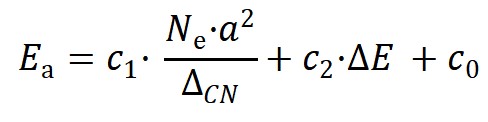
其中Ne為金屬催化劑的價電子數����,a為相應的晶格常數,ΔE為反應熱�,c1,c2和c0是對應的系數��。在該二維模型中,第一項為結構項�,正比于價電子數和晶格常數平方大小���,反比于拓撲配位不飽和度��,第二項為經典BEP關系中的反應能項。該模型能夠準確預測分子斷鍵能壘�,并在具有不同對稱性��、鍵級和空間位阻等化學鍵的數據集上表現出良好的普適性(圖2b和2c)。
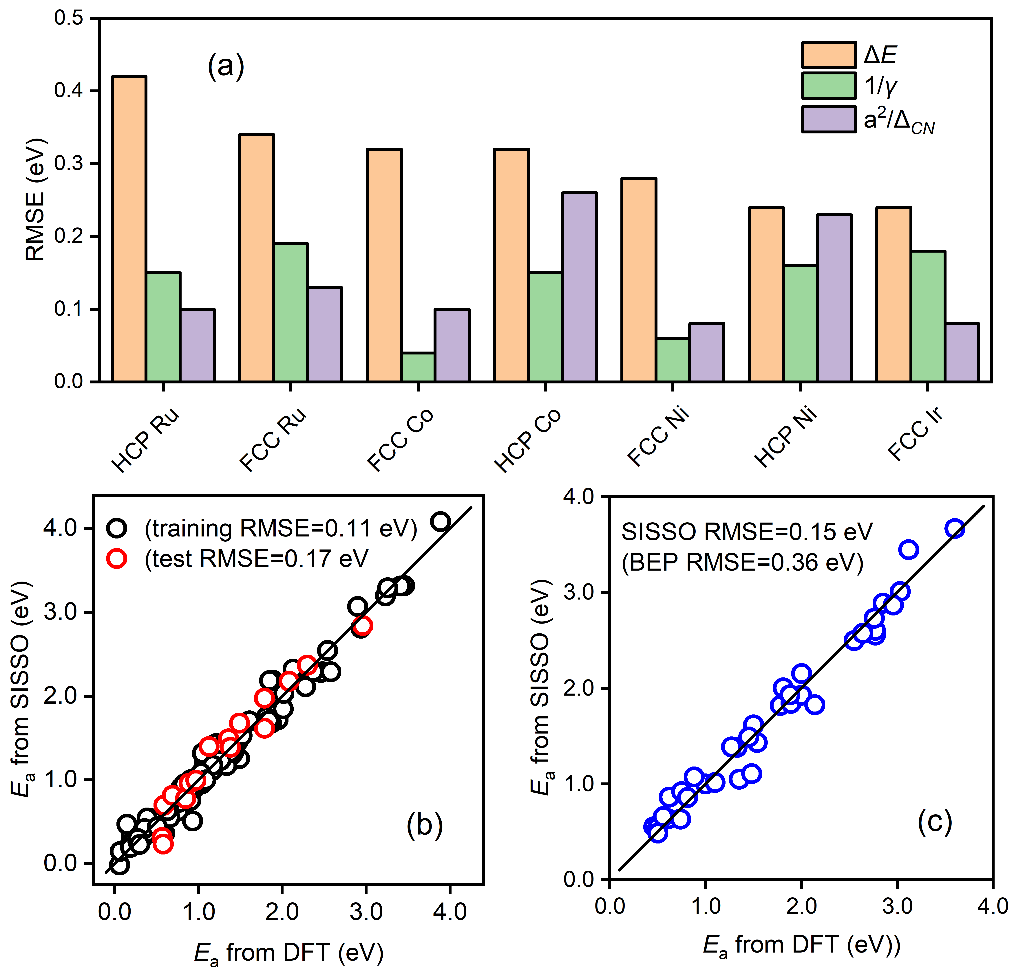
圖2 (a)CO活化能壘與不同物理參數(反應熱���、表面能、拓撲配位不飽和度)之間的統計相關性�;(b)機器學習模型的訓練和測試準確度�;(c)模型在新體系上的遷移性預測能力
上述方程由于明確包含了催化劑的組分�����、結構和反應熱信息���,因此活化能對組分調制的結構項(圖3a)和能量項(圖3b)的依賴關系可用來拆分催化劑的幾何效應和電子效應�。同時活化能對這兩項投影的大小可用來對催化結構敏感性進行分類�。如圖3c所示,活化能在前者較大的投影和系數意味著該分子(比如CO, NO, N2)的活化過程是一種結構敏感的反應���,而如果在后者上(比如OH�����,NH)則意味著該反應為結構不敏感�����,這一結論主要是適合于小分子���。較大的分子因其空間位阻效應顯著��,相應的投影不能用來判斷反應的結構敏感性,但相應公式的預測能力依然表現出色���。
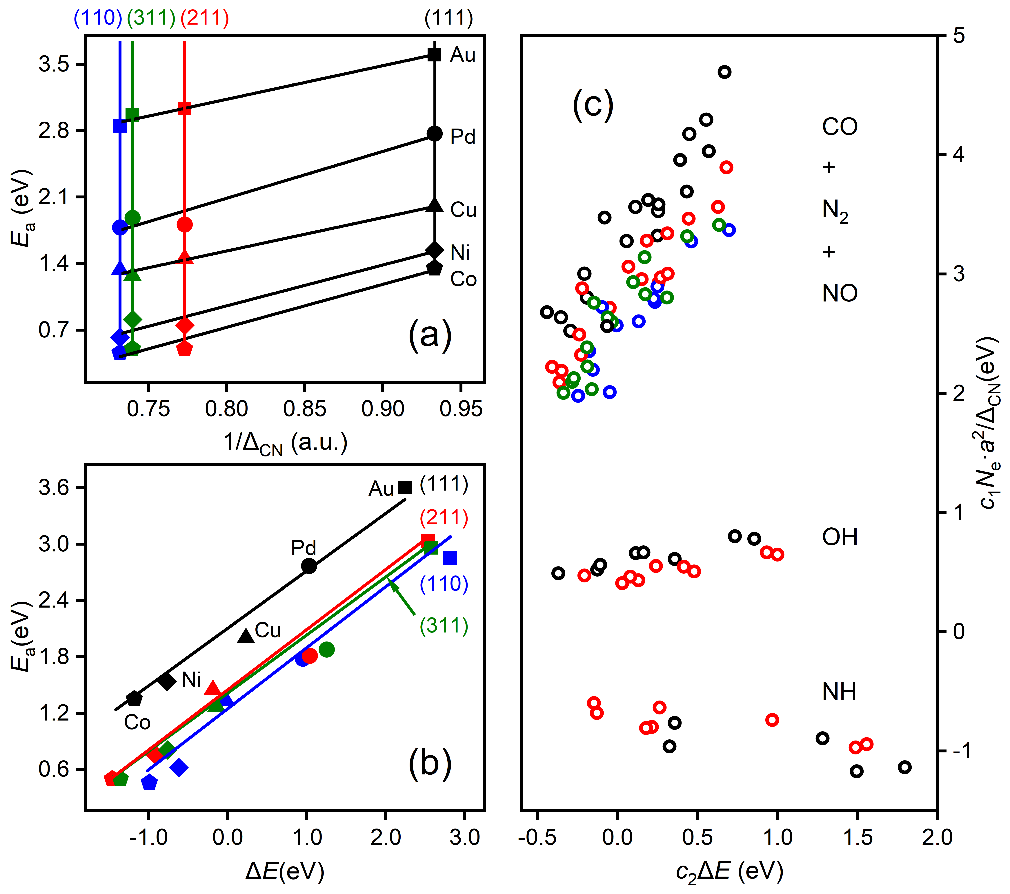
圖3 活化能壘的幾何效應與電子效應、結構效應與能量效應的去糾纏統計分析
中國科大博士生舒武為第一作者�����,中國科大的李微雪教授為通訊作者�����,上海大學的歐陽潤海博士為共同通訊作者�。該項工作得到了國家自然科學基金創新研究群體���、面上項目����、中國科學院先導項目、科技部項目資助,以及中國科學技術大學超算中心為該項目研究提供的機時����。
文章鏈接:https://pubs.acs.org/doi/10.1021/jacs.4c01524
(中國科學技術大學化學與材料科學學院,精準智能化學重點實驗室、合肥微尺度物質科學國家研究中心�,科研部)

