近日,中國科學技術大學生命科學與醫學部瞿昆教授課題組、北京生命科學研究所黎斌研究員課題組,以及中國科學技術大學數學科學學院陳發來教授課題組聯合完成了一項大規模研究。他們通過對百萬量級單細胞多組學數據進行分析,系統評估了14種單細胞模態預測算法和18種單細胞多組學整合算法的性能。該研究成果以題為“Benchmarking algorithms for single-cell multi-omics prediction and integration”的論文,于2024年9月25日在線發表于國際知名學術期刊《Nature Methods》。

單細胞多組學技術(如CITE-seq、REAP-seq、SHARE-seq和10x Multiome等)的發展,為深入理解細胞功能和復雜的基因調控機制提供了前所未有的機遇。然而,濕實驗方法通常伴隨高成本、數據質量有限以及批次效應等挑戰。為克服這些局限,生物信息學家基于統計模型和人工智能技術,開發了多種算法。這些算法不僅能夠利用單細胞轉錄組數據推斷同一細胞內的蛋白質豐度和染色質可及性信息,還通過將不同模態的數據映射到統一的特征空間實現數據整合,去除批次效應。這些工具大大提升了現有單細胞數據的解析能力。然而,面對海量數據和眾多算法,研究人員往往難以判斷哪些工具最適合他們的研究,因此,對這些算法進行基準測試(benchmarking)尤為重要。
在本次研究中,團隊收集了來自47個數據集的上百萬個單細胞多組學數據,涵蓋多個生物樣本和實驗平臺。他們設計了一套全面的評估流程,結合算法的準確性、魯棒性和計算資源消耗等多維度指標,系統評估了領域內最常用的算法。結果顯示,在蛋白質豐度預測方面,totalVI和scArches表現最為優異;在染色質可及性預測中,LS_Lab算法排名領先。在多組學整合分析中,Seurat、MOJITOO和scAI在垂直整合上表現突出,而totalVI和UINMF在水平整合和馬賽克整合任務中展現了卓越性能。這一研究不僅為算法設計提供了新思路,還為未來多組學數據的分析和應用奠定了重要基礎。為幫助科研人員選擇合適的分析工具,研究團隊在GitHub上發布了完整的分析流程、代碼和測試數據集,供同行使用和改進。
研究團隊還通過深入探討這些算法的數學原理,發現降噪處理是提高單細胞數據預測精度的關鍵。在性能評估中,機器學習算法(如基于奇異值分解的LS_Lab和Guanlab-dengkw)以及基于概率模型的深度學習算法(如totalVI)均表現出顯著優勢。然而,研究還指出,現有模態預測算法在某些關鍵蛋白的預測性能上仍有待提升,染色質可及性預測的準確性也需進一步優化。
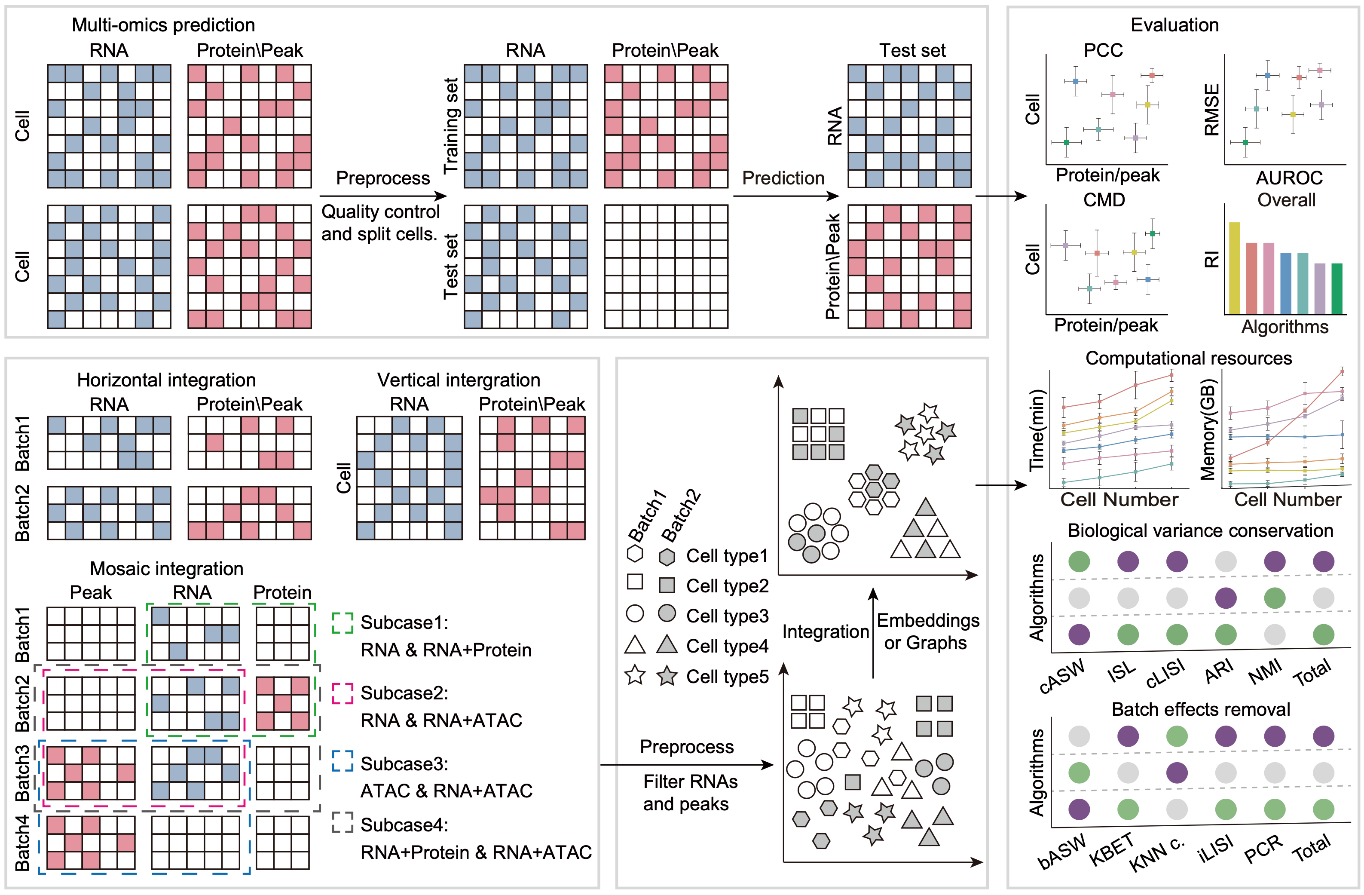
圖.評估流程示意圖
該研究由瞿昆教授、黎斌研究員和陳發來教授共同指導并擔任通訊作者,博士后胡銀雷、博士生萬思遠和羅袁涵宇為共同第一作者。該研究得到了國家自然科學基金、科技部重點研發專項等多項資助,中國科學技術大學超級計算中心及生命科學學院生物信息學中心為項目提供了關鍵計算資源支持。
在組學大數據時代,對復雜數據的精確解析需要依賴生物學與數學、計算機科學的深度融合。跨學科合作不僅推動了生物醫學領域的創新發展,也為未來研究提供了新的可能性。此次研究的成功正是多學科背景團隊密切合作的結果,充分展示了學科交叉在現代生物學研究中的重要性。通過這樣的合作,研究團隊期望進一步推動單細胞多組學技術在科學研究中的廣泛應用,為基礎研究和臨床應用提供新的洞見。
論文鏈接:https://www.nature.com/articles/s41592-024-02429-w
(生命科學與醫學部、數學科學學院、科研部)

